CRISPR/Cas9 基因剔除技術
一、什麼是 CRISPR/Cas9 ?
一個自細菌中所發現的免疫系統將造成基因組編輯工具產生翻天覆地的大躍進。在遭受如病毒或噬菌體的感染後,其實原核生物例如細菌也是會生病。這個問題對於食物工業產生的衝擊是很大的,例如嗜熱鏈球菌 (Streptococcus thermophilus) 是主要是用來產生優酪乳或起士。當嗜熱鏈球菌受到噬菌體的感染將嚴重減少優酪乳與起士的產量。因此在 2007 年杜邦公司 (DuPont) 發現了一種方法來解決嗜熱鏈球菌受噬菌體感染的問題 [1],也就是 CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats) 系統。在 2013 年2月開始, 研究人員陸續將 CRISPR 進行改造,讓此工具可以有效辨識與剪切
特定的 DNA 序列,並成功將此技術應用於細胞、小鼠、大鼠、斑馬魚、細菌、果蠅、酵母菌、線蟲或農作物中進行基因組編輯工作。CRISPR 分為三種類型。第一型是透過 Cascade:crRNA 複合體去辨識互補的 DNA ,再經由 Cas3 核酸酶進行 DNA 降解。而第三型與第一型較為類似,一個包含 Csm 或 Cmr 蛋白與 Cas6 蛋白的複合體與 pre-crRNA 結合並且加
工處理pre-crRNA 形成 crRNA ,透過這樣複雜的複合體來辨識與降解互補的DNA 序列。然而第二型的 CRISPR/Cas9 系統無須產生一個複雜的蛋白複合體,僅透過 Cas9 蛋白結合由 crRNA 與 tracrRNA 形成的 pre-crRNA 就可以讓RNase III 加工形成成熟的 crRNA ,使得 Cas9/ tracrRNA:crRNA 複合體可以去辨識與切割互補的 DNA 。由於第二型 CRISPR 系統的簡易性與方便性,因此於 2013 年被開發並廣泛應用在各種不同物種的基因剔除研究上。
雖然第二型 CRISPR/Cas9 系統相較於其他兩類型來說,沒有複雜的蛋白複合體結構顯得簡單許多,但是仍須具備 crRNA 與 tracrRNA 才能進行 DNA辨識,因此Martin Jinek 與 Krzysztof Chylinski 等人嘗試將 crRNA 與tracrRNA 相接形成嵌合體 (single chimeric RNA or guide RNA) 並且成功保留其 DNA 辨識與切割的活性。後來經過Woong Y Hwang 等人改良延長
tracrRNA 的三端序列則可有效在斑馬魚胚胎中進行基因標靶剔除。
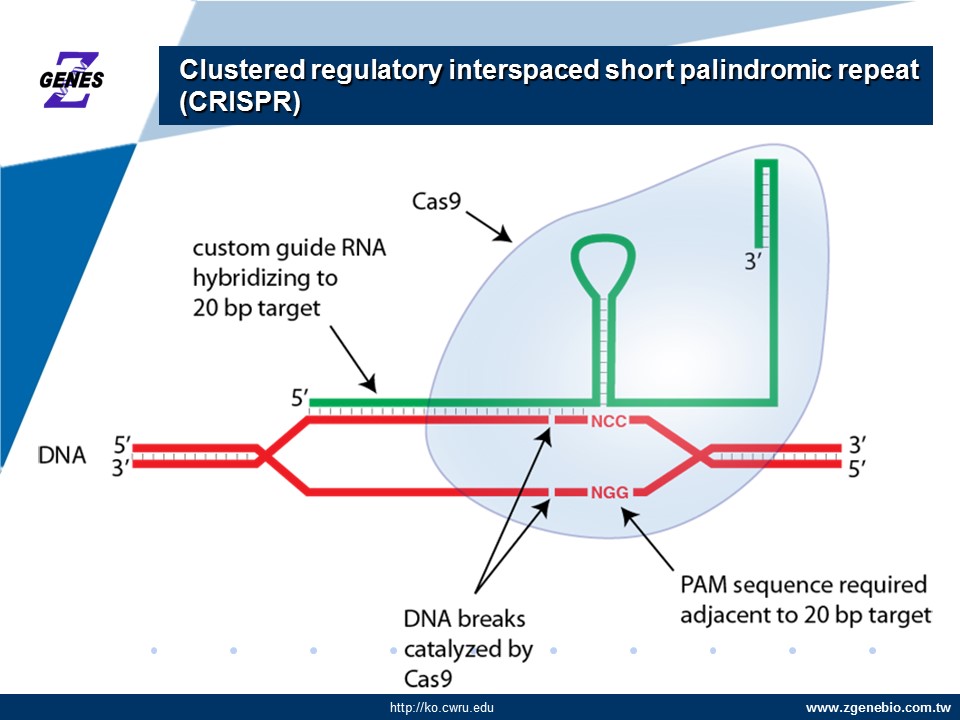
第二型 CRISPR/Cas9 應用在基因工程上,主要透過一個來自於化膿性鏈球菌 (Streptococcus pyogenes) 的 Cas9 蛋白與一個 gRNA 形成一個複合體,該複合體會與互補的 DNA 進行辨識。辨識 DNA 序列最主要的是透過 gRNA前端 (5 端) 的 20 nt 長度的序列,稱之為 Protospacer 。緊接在後的三個核苷酸序列 (NGG) 則稱之為 Protospacer adjacent motif (PAM),主要是讓 Cas9 辨識並且切割 DNA。目前發現在細胞實驗中,野生型Cas9 存在較高的脫靶可能性,主要原因是當 Protospacer 的5 端序列約5-7 bps 的核苷酸產生變異時,野生型的Cas9 蛋白仍然可以對 DNA 進行辨識與切割。但是對於動物胚胎打靶時,由於脫靶嚴重的胚胎將無法正常發育,因此篩選到的動物個體脫靶問題較不明顯。
一個自細菌中所發現的免疫系統將造成基因組編輯工具產生翻天覆地的大躍進。在遭受如病毒或噬菌體的感染後,其實原核生物例如細菌也是會生病。這個問題對於食物工業產生的衝擊是很大的,例如嗜熱鏈球菌 (Streptococcus thermophilus) 是主要是用來產生優酪乳或起士。當嗜熱鏈球菌受到噬菌體的感染將嚴重減少優酪乳與起士的產量。因此在 2007 年杜邦公司 (DuPont) 發現了一種方法來解決嗜熱鏈球菌受噬菌體感染的問題 [1],也就是 CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats) 系統。在 2013 年2月開始, 研究人員陸續將 CRISPR 進行改造,讓此工具可以有效辨識與剪切
特定的 DNA 序列,並成功將此技術應用於細胞、小鼠、大鼠、斑馬魚、細菌、果蠅、酵母菌、線蟲或農作物中進行基因組編輯工作。CRISPR 分為三種類型。第一型是透過 Cascade:crRNA 複合體去辨識互補的 DNA ,再經由 Cas3 核酸酶進行 DNA 降解。而第三型與第一型較為類似,一個包含 Csm 或 Cmr 蛋白與 Cas6 蛋白的複合體與 pre-crRNA 結合並且加
工處理pre-crRNA 形成 crRNA ,透過這樣複雜的複合體來辨識與降解互補的DNA 序列。然而第二型的 CRISPR/Cas9 系統無須產生一個複雜的蛋白複合體,僅透過 Cas9 蛋白結合由 crRNA 與 tracrRNA 形成的 pre-crRNA 就可以讓RNase III 加工形成成熟的 crRNA ,使得 Cas9/ tracrRNA:crRNA 複合體可以去辨識與切割互補的 DNA 。由於第二型 CRISPR 系統的簡易性與方便性,因此於 2013 年被開發並廣泛應用在各種不同物種的基因剔除研究上。
雖然第二型 CRISPR/Cas9 系統相較於其他兩類型來說,沒有複雜的蛋白複合體結構顯得簡單許多,但是仍須具備 crRNA 與 tracrRNA 才能進行 DNA辨識,因此Martin Jinek 與 Krzysztof Chylinski 等人嘗試將 crRNA 與tracrRNA 相接形成嵌合體 (single chimeric RNA or guide RNA) 並且成功保留其 DNA 辨識與切割的活性。後來經過Woong Y Hwang 等人改良延長
tracrRNA 的三端序列則可有效在斑馬魚胚胎中進行基因標靶剔除。
第二型 CRISPR/Cas9 應用在基因工程上,主要透過一個來自於化膿性鏈球菌 (Streptococcus pyogenes) 的 Cas9 蛋白與一個 gRNA 形成一個複合體,該複合體會與互補的 DNA 進行辨識。辨識 DNA 序列最主要的是透過 gRNA前端 (5 端) 的 20 nt 長度的序列,稱之為 Protospacer 。緊接在後的三個核苷酸序列 (NGG) 則稱之為 Protospacer adjacent motif (PAM),主要是讓 Cas9 辨識並且切割 DNA。目前發現在細胞實驗中,野生型Cas9 存在較高的脫靶可能性,主要原因是當 Protospacer 的5 端序列約5-7 bps 的核苷酸產生變異時,野生型的Cas9 蛋白仍然可以對 DNA 進行辨識與切割。但是對於動物胚胎打靶時,由於脫靶嚴重的胚胎將無法正常發育,因此篩選到的動物個體脫靶問題較不明顯。
|
|
二、 CRISPR/Cas9 的優點為何?
1. 無物種限制,可在動物、植物、細菌等生物中進行基因打靶。
2. 載體構築簡單快速,幾天內即可完成構築,失敗率低。
3. 靶位點設計容易,PAM 位點 (NGG) 於 128 bp 隨機 DNA 序列中就會出現一次,並且正反兩股皆可設計。
4. Cas9 辨識 DNA 序列效率高,新開發的 Cas9 D10A 可有效降低脫靶效應,增加打靶準確性。
5. 基因打靶成功率較 ZFN 或 TALEN 高,還可同時放入兩個以上的sgRNA 來進行多個基因同時剔除的目的。
6. 雙合一或三合一載體簡化細胞轉染的難度。搭配多種報導基因,如螢光蛋白或抗生素篩選基因,可使實驗設計更有彈性。
7. 可以直接作用於 DNA 上,讓基因默化更容易,將可漸漸取代 RNA(RNAi) 或 Morpholino 等基因干擾技術。
1. 無物種限制,可在動物、植物、細菌等生物中進行基因打靶。
2. 載體構築簡單快速,幾天內即可完成構築,失敗率低。
3. 靶位點設計容易,PAM 位點 (NGG) 於 128 bp 隨機 DNA 序列中就會出現一次,並且正反兩股皆可設計。
4. Cas9 辨識 DNA 序列效率高,新開發的 Cas9 D10A 可有效降低脫靶效應,增加打靶準確性。
5. 基因打靶成功率較 ZFN 或 TALEN 高,還可同時放入兩個以上的sgRNA 來進行多個基因同時剔除的目的。
6. 雙合一或三合一載體簡化細胞轉染的難度。搭配多種報導基因,如螢光蛋白或抗生素篩選基因,可使實驗設計更有彈性。
7. 可以直接作用於 DNA 上,讓基因默化更容易,將可漸漸取代 RNA(RNAi) 或 Morpholino 等基因干擾技術。
TALEN 和 CRISPR 比較
Relative characteristics of genome-editing tools
Relative characteristics of genome-editing tools

TALEN
實驗室組裝較難,但Off-target機率極低,經過我們改造的第
三代高活性TALEN效率很好
CRISPR
操作作較簡單,但有Off-target 問題,但操作簡單 效率高 許多實驗室採用這系統
Q:如何降低Off-target
1.用短版Target site設計:18nt
2.可以選擇D10A載體
3.選擇最新Cas9 1.1版本載體
三、 如何靈活運用 CRISPR/Cas9 基因剔除技術?
在 1980 年末,開發了一種透過同源互換 (Homologous recombination, HR)方式將特定基因突變或定點刪除 DNA 的技術。這個技術主要是透過設計一段相似的 DNA 片段來置換染色體上的一段 DNA 。然而,在自然狀況下,體細胞要發生同源互換的機率其實是非常低的,通常低於 1*10-6。這樣的技術較適合應用於幹細胞或胚幹細胞,但是對於胚幹細胞尚未開發成功的物種而言,想要利用同源互換方式進行基因剔除卻是非常困難的事情。但是隨著時代的進步,漸漸地衍生出 RNAi 或 Morpholino 基因干擾技術,但是還是無法滿足研究或臨床的需要。所以在特定 DNA 位點上進行 DSB 是研究人員一直想要達成的目標。近年來,分子生物技術的成熟,漸漸開發出許多新的技術,如:Cre/LoxP 、 ZFN 、 TALEN 與最新的 CRISPR 系統可以在定點進行 DNA
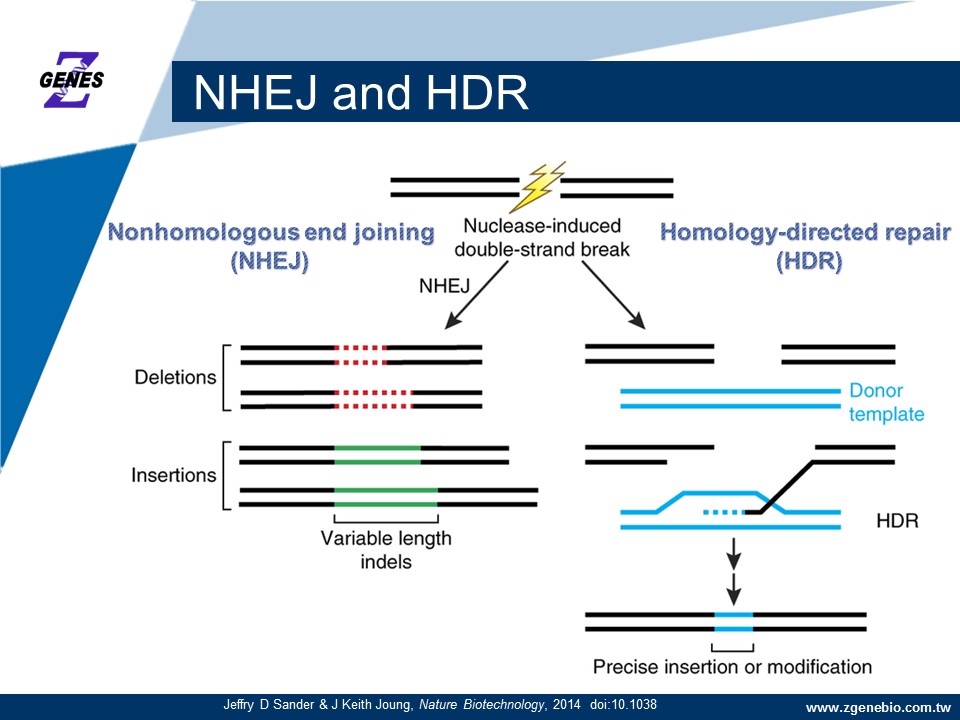
編輯。目前許多研究人員對於這個沒有物種限制的 CRISPR/Cas9 技術感到興趣,只要能成功將 Cas9 mRNA/sgRNA 或 CRISPR/Cas9 多合一載體送入細胞或胚胎中,即可進行基因剔除實驗。在細胞中, Cas9 蛋白與 sgRNA 形成複合體,並且對於 DNA 序列進行辨認並解開雙股 DNA 結構。因此, crRNA 與DNA 單股進行雜交並且透過 Cas9 蛋白 N 端的 RuvC 與蛋白中部的 HNH這兩個活性區作用將 DNA 雙股切斷而形成雙股 DNA 斷裂 (Double strandbreak, DSB)。當細胞察覺到 DSB 時,啟動非同源互換型 DNA 修復機制(Non-homologous end joining, NHEJ) 或進行同源互換 DNA 修復機制,這個過程往往造成部分 DNA 缺失或插入讓基因編碼產生位移,最後造成基因破壞。在技術上的演進, CRISPR/Cas9 更可能衍生出更多的實驗策略,
如:1) 可進行基因剔除工作 : CRISPR/Cas9 導致 DNA 的雙股斷裂,誘發NHEJ 修復機制,結果引起無法預測的 DNA 片段刪除或插入 (Indel)
編輯。目前許多研究人員對於這個沒有物種限制的 CRISPR/Cas9 技術感到興趣,只要能成功將 Cas9 mRNA/sgRNA 或 CRISPR/Cas9 多合一載體送入細胞或胚胎中,即可進行基因剔除實驗。在細胞中, Cas9 蛋白與 sgRNA 形成複合體,並且對於 DNA 序列進行辨認並解開雙股 DNA 結構。因此, crRNA 與DNA 單股進行雜交並且透過 Cas9 蛋白 N 端的 RuvC 與蛋白中部的 HNH這兩個活性區作用將 DNA 雙股切斷而形成雙股 DNA 斷裂 (Double strandbreak, DSB)。當細胞察覺到 DSB 時,啟動非同源互換型 DNA 修復機制(Non-homologous end joining, NHEJ) 或進行同源互換 DNA 修復機制,這個過程往往造成部分 DNA 缺失或插入讓基因編碼產生位移,最後造成基因破壞。在技術上的演進, CRISPR/Cas9 更可能衍生出更多的實驗策略,
如:1) 可進行基因剔除工作 : CRISPR/Cas9 導致 DNA 的雙股斷裂,誘發NHEJ 修復機制,結果引起無法預測的 DNA 片段刪除或插入 (Indel)
NHEJ AND HDR
2) 可進行基因大片段剔除工作 : 當 DNA 上設計兩個相鄰的CRISPR/Cas9 靶位點,透過兩個位點的切割,有機會造成一個較大 DNA片段的移除工作。
3) 可進行多基因同時剔除工作 : 若同時對細胞轉染或胚胎注射多個位點的sgRNAs 時,有機會可以同時將多個基因做一次性的破壞工作,形成double 甚至是triple mutants。
4) 可進行基因點突變置換工作 : 同時存在有同源性的單股 DNA (Singlestranded Oligo, ssOligo) 即可透過同源互換方式將帶有單點核苷酸錯誤的序列置換原本的 DNA 序列。相同的,透過 CRISPR/Cas9 或CRISPR/Cas9 D10A 系統皆可以達成此目的。此策略的優點在於同源互換臂的長度僅需要 18-20 nt 左右即可,但是缺點在於欲插入的序列長度不長(<50 nt) 。
5) 可進行基因同源互換與基因敲入工作 : 當欲插入 DNA 序列大於 50 nt時,此時將無法再透過 ssOligo 進行同源互換,所以必須要構築一個Donor vector 來進行HR。當欲插入 DNA 序列長度越長時,可以透過增加左右同源互換臂 (>500 bp) 的長度來增加其成功機率。此方法優點在於可以放入1 至6 kb 的 DNA 序列,因此可以包含一個小型啟動子與報導基因等等。
6) 可進行染色體轉位與基因融合研究 : 透過定點打靶可以用來研究染色體位移 (Chromosomal translocations) 的疾病。某些疾病的成因是因為染色體位移而產生特殊的融合蛋白基因而形成致癌基因。所以在特定的兩個基因上設計 CRISPR/Cas9 靶位點後,由於 DNA 雙股斷裂造成染色體位移以及產生融合基因的研究已經被證實,對於染色體位移相關實驗是非常重要的突破。
7) 可在轉錄層次去干擾基因的表現 : 最近研究指出,Cas9 D10A 與 Cas9H840A 雙重突變會造成Cas9 蛋白質完全失去 DNA 切割能力,但是仍保留 Cas9 與 sgRNA 結合與辨識 DNA 的功能。若經過重組將帶有D10A 與 H840A 雙重突變的 Cas9 蛋白與 VP64 活化子或抑制子進行融合,這樣的重組蛋白就可以定點結合某基因啟動子區域的 DNA 以及調高或抑制該基因表現,稱之為 CRISPR 干擾技術 (CRISPR interference,CRISPRi)。
3) 可進行多基因同時剔除工作 : 若同時對細胞轉染或胚胎注射多個位點的sgRNAs 時,有機會可以同時將多個基因做一次性的破壞工作,形成double 甚至是triple mutants。
4) 可進行基因點突變置換工作 : 同時存在有同源性的單股 DNA (Singlestranded Oligo, ssOligo) 即可透過同源互換方式將帶有單點核苷酸錯誤的序列置換原本的 DNA 序列。相同的,透過 CRISPR/Cas9 或CRISPR/Cas9 D10A 系統皆可以達成此目的。此策略的優點在於同源互換臂的長度僅需要 18-20 nt 左右即可,但是缺點在於欲插入的序列長度不長(<50 nt) 。
5) 可進行基因同源互換與基因敲入工作 : 當欲插入 DNA 序列大於 50 nt時,此時將無法再透過 ssOligo 進行同源互換,所以必須要構築一個Donor vector 來進行HR。當欲插入 DNA 序列長度越長時,可以透過增加左右同源互換臂 (>500 bp) 的長度來增加其成功機率。此方法優點在於可以放入1 至6 kb 的 DNA 序列,因此可以包含一個小型啟動子與報導基因等等。
6) 可進行染色體轉位與基因融合研究 : 透過定點打靶可以用來研究染色體位移 (Chromosomal translocations) 的疾病。某些疾病的成因是因為染色體位移而產生特殊的融合蛋白基因而形成致癌基因。所以在特定的兩個基因上設計 CRISPR/Cas9 靶位點後,由於 DNA 雙股斷裂造成染色體位移以及產生融合基因的研究已經被證實,對於染色體位移相關實驗是非常重要的突破。
7) 可在轉錄層次去干擾基因的表現 : 最近研究指出,Cas9 D10A 與 Cas9H840A 雙重突變會造成Cas9 蛋白質完全失去 DNA 切割能力,但是仍保留 Cas9 與 sgRNA 結合與辨識 DNA 的功能。若經過重組將帶有D10A 與 H840A 雙重突變的 Cas9 蛋白與 VP64 活化子或抑制子進行融合,這樣的重組蛋白就可以定點結合某基因啟動子區域的 DNA 以及調高或抑制該基因表現,稱之為 CRISPR 干擾技術 (CRISPR interference,CRISPRi)。
Q:我們可以控制突變的缺失嗎,要如何挑選突變品系?
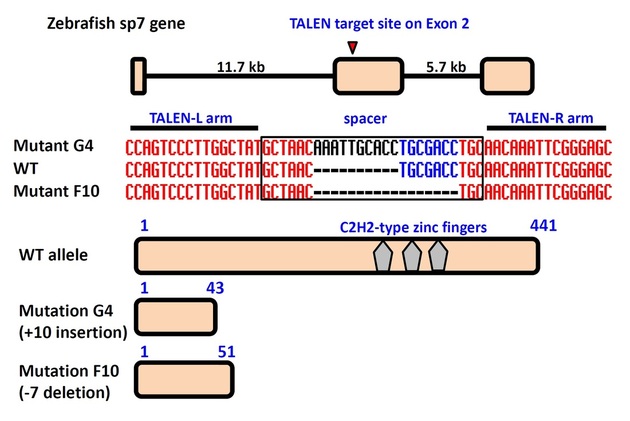
CRISPR 技術可準確的找到我們設計特定序列利用Cas9產生雙股斷列,但無法預知修補後的缺失,
修補過程會隨機產生DNA序列產生Indel 需利用定序去確認突變片段,序列產生缺失我們建議選擇
避開3的倍數,建立多種突變品系,如圖示我們選擇G4 & F10 品系成功造成蛋白質終止,產生突變
Q:突變產生是Heterozygous還是Homozygous??若是細胞株要如何產生homo細胞株??
一般大部份機率會產生Heterozygous只有單一染色體產生突變只有很小的機率產生Homozygous,若只有挑到heter F1 可以利用一對heter F1 進行交配產生homo突變株
若是細胞株部份我們會建立heter細胞系,再進行一次轉染即可以大大提升homo突變的機率
CRISPR 在植物應用
CRISPR 已成功應用在水稻,阿拉伯芥(擬南芥),菸草,蕃茄等物種,利用農桿菌將CRISPR質體送入植物
植物操作需注意多倍體,三倍體的剔除機率會比二倍體低很多
我們有專們為植物農桿菌轉殖設計專門的植物載體,可幫助實驗室輕鬆上手,無須再改造載體
四、力鈞生物公司 CRISPR 快速構築試劑盒的優點
力鈞生技公司目前推出的『快速CRISPR 構建試劑盒』具備以下特點:
1) 簡單:只需單一步驟合成,失敗率低,大學部學生即可勝任。整個載體構築流程非常簡單,無任何繁瑣步驟。
2) 快速: 只要配合本公司優化的載體構築步驟,只需2 天時間就能完成CRISPR 質體構建。
3) 費用省:組裝經費、人力和時間成本低。平均自行組裝一個 CRISPR 載體只需要台幣兩千元左右的花費。
4) 選擇性多:具有多種載體骨架可供選擇,方便進行細胞或動物基因剔除實驗使用。
5) 產品持續更新:對應不同的實驗需求,力鈞公司持續開發新載體供客戶選擇。
1) 簡單:只需單一步驟合成,失敗率低,大學部學生即可勝任。整個載體構築流程非常簡單,無任何繁瑣步驟。
2) 快速: 只要配合本公司優化的載體構築步驟,只需2 天時間就能完成CRISPR 質體構建。
3) 費用省:組裝經費、人力和時間成本低。平均自行組裝一個 CRISPR 載體只需要台幣兩千元左右的花費。
4) 選擇性多:具有多種載體骨架可供選擇,方便進行細胞或動物基因剔除實驗使用。
5) 產品持續更新:對應不同的實驗需求,力鈞公司持續開發新載體供客戶選擇。

五、該如何設計 CRISPR 靶位點?
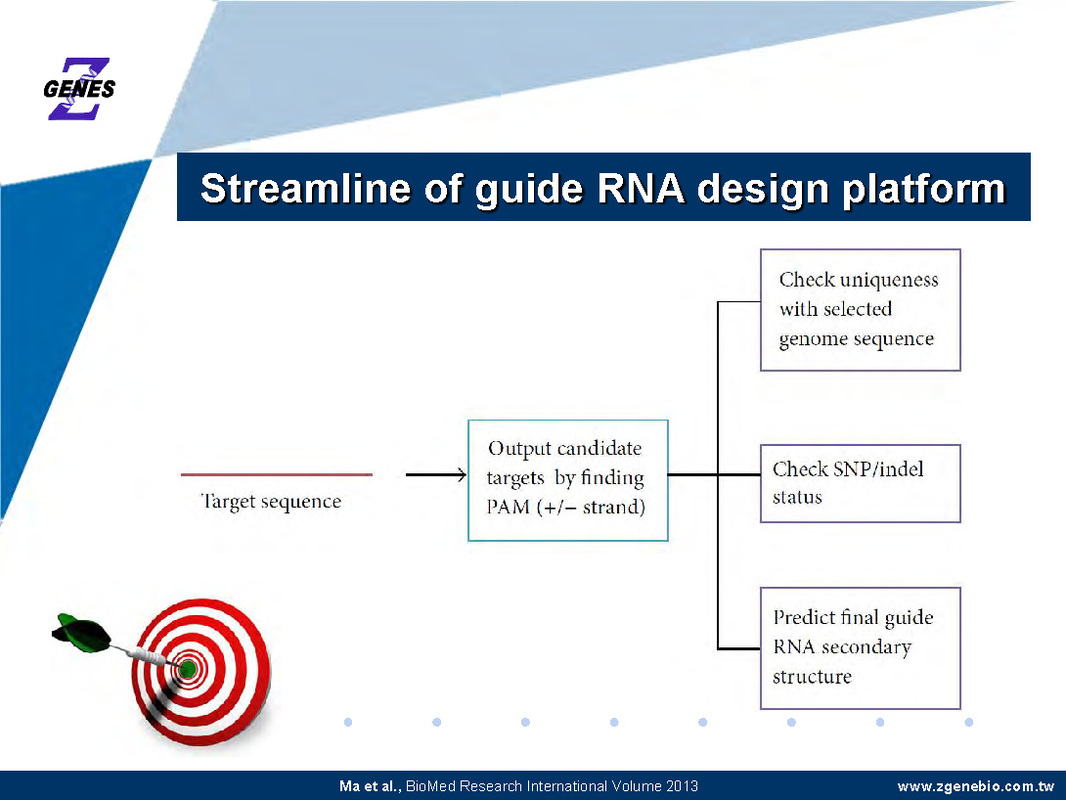
1.進入 Ensembl 網頁 (http://www.ensembl.org/index.html) 搜尋要標定的基因。
2.E-CRISP 網頁 (http://www.e-crisp.org/E-CRISP/)。選擇 De-novo或點選圖片來設計新靶位點。
3.選擇物種並貼上 Accession Number,另外也可以選擇貼上 FASTA Format格式的序列。
4.選擇靶位點設計模式。標準 Cas9 靶位點設計選 Single design,若要設計 Nickase 靶位點,請選擇 Paired designs。若有特殊需求可以勾選Select if CRISPR designs should be used for tagging or knockout experiments,並且選擇需要的模式。另外,若想修改設計靶位點的參數,可以點選click here to change CRISPR properties 去展開參數設定選項。
5.條件過濾設定,建議勾選前三項
6.設定 Off-target site 分析參數。下圖所示參數較為寬鬆,以免找不到適合的靶位點。另外,右側 Select to check for secondary off-targets 則可以進行特定篩選基因或特定序列進行預測。
7.結果輸出設定。可限制每個 Exon 顯示多少個靶位點。當設定完成即可點選 Start 進行分析
8.結果輸出網頁,序列一一列出於表單中,即可依照需求選擇適合的靶位點。
2.E-CRISP 網頁 (http://www.e-crisp.org/E-CRISP/)。選擇 De-novo或點選圖片來設計新靶位點。
3.選擇物種並貼上 Accession Number,另外也可以選擇貼上 FASTA Format格式的序列。
4.選擇靶位點設計模式。標準 Cas9 靶位點設計選 Single design,若要設計 Nickase 靶位點,請選擇 Paired designs。若有特殊需求可以勾選Select if CRISPR designs should be used for tagging or knockout experiments,並且選擇需要的模式。另外,若想修改設計靶位點的參數,可以點選click here to change CRISPR properties 去展開參數設定選項。
5.條件過濾設定,建議勾選前三項
6.設定 Off-target site 分析參數。下圖所示參數較為寬鬆,以免找不到適合的靶位點。另外,右側 Select to check for secondary off-targets 則可以進行特定篩選基因或特定序列進行預測。
7.結果輸出設定。可限制每個 Exon 顯示多少個靶位點。當設定完成即可點選 Start 進行分析
8.結果輸出網頁,序列一一列出於表單中,即可依照需求選擇適合的靶位點。
我們提供 力鈞免費靶點設計服務

|

|
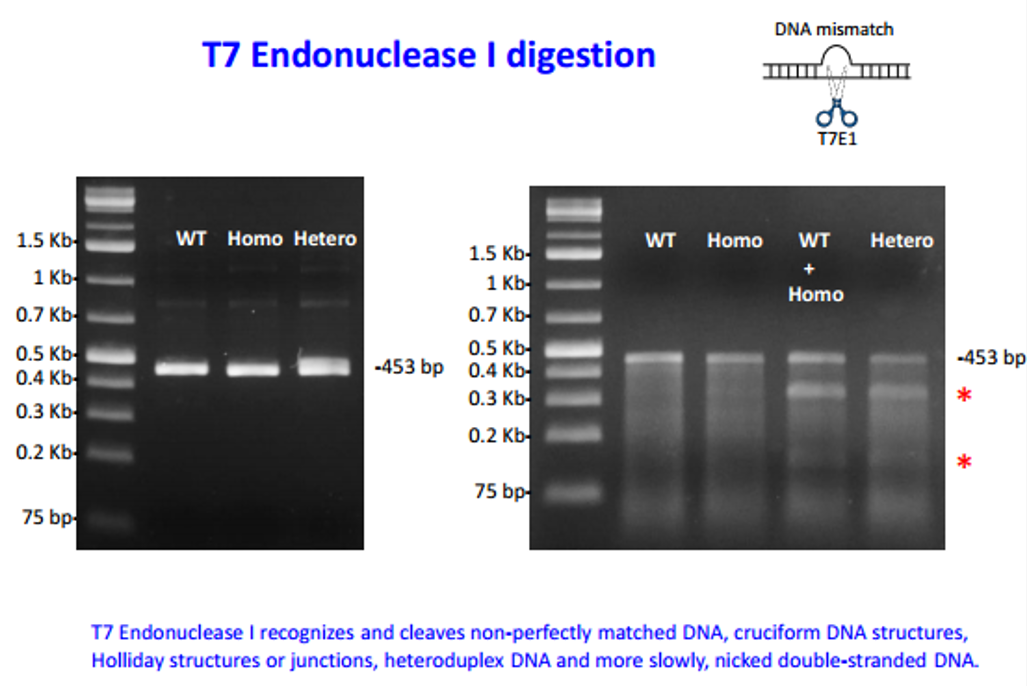
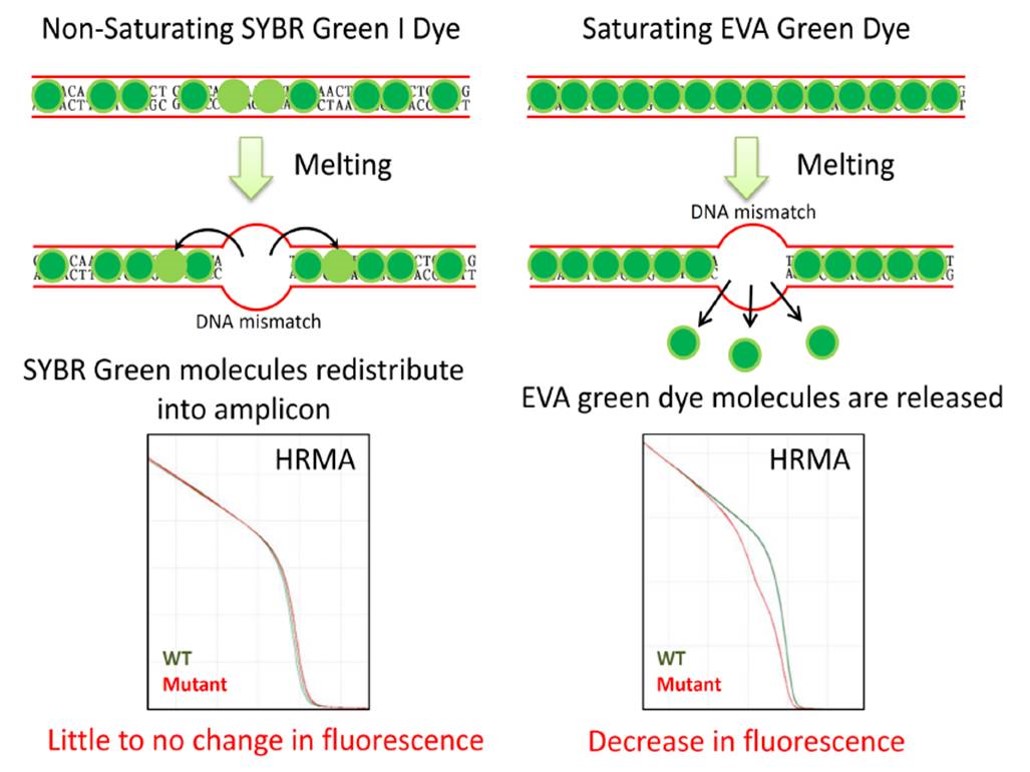
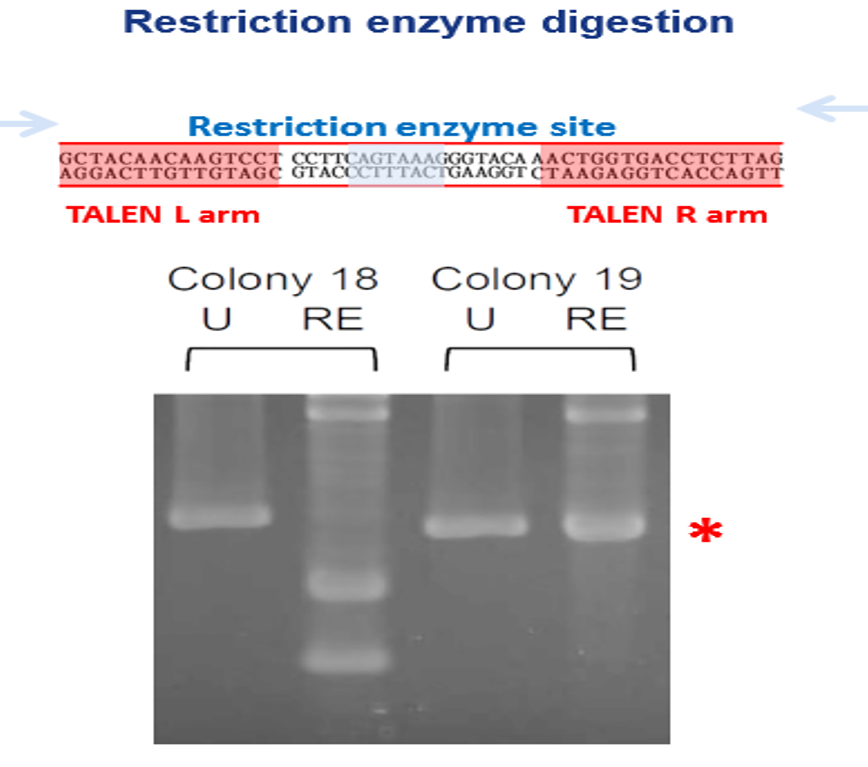
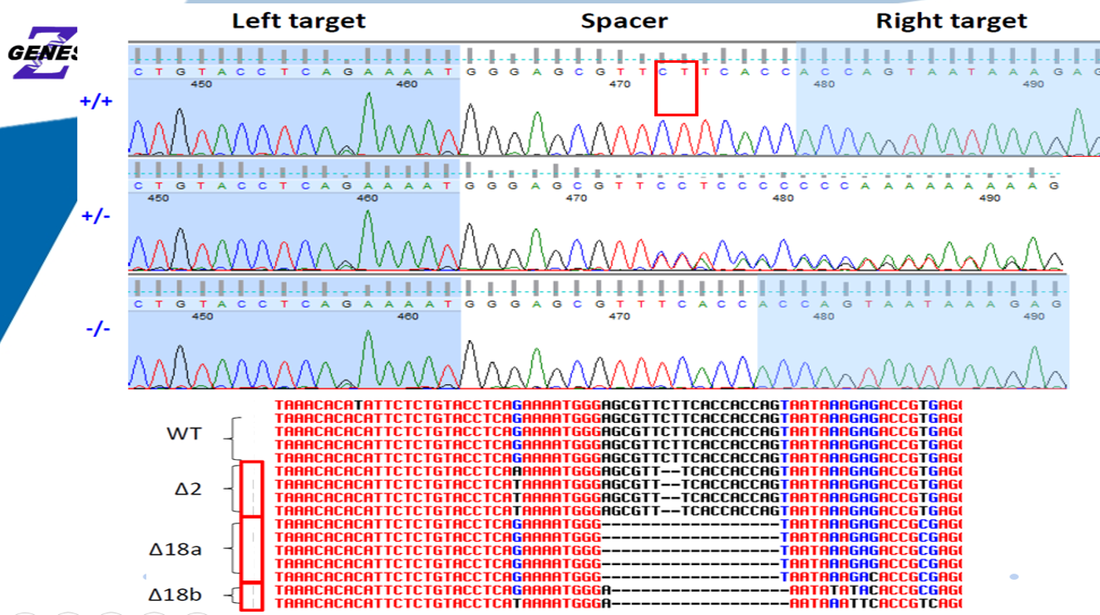
突變篩選方法
1.利用T7E1進行篩選
2.利用HRM高熔點分析進行篩選
3.利用限制酵素
4.利用定序直接篩選
|
|
|
定序
|
參考文獻
1. Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, et al.(2007) CRISPR provides acquired resistance against viruses in prokaryotes.
Science 315: 1709-1712.
2. Richter H, Randau L, Plagens A (2013) Exploiting CRISPR/Cas:interference mechanisms and applications. Int J Mol Sci 14: 14518-14531.
3. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, et al. (2012) Aprogrammable dual-RNA-guided DNA endonuclease in adaptive bacterial
immunity. Science 337: 816-821.
4. Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, et al. (2013)Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31: 227-229.
5. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, et al. (2013)High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31: 822-826.
6. Cradick TJ, Fine EJ, Antico CJ, Bao G (2013) CRISPR/Cas9 systemstargeting beta-globin and CCR5 genes have substantial off-target activity.Nucleic Acids Res 41: 9584-9592.
7. Hruscha A, Krawitz P, Rechenberg A, Heinrich V, Hecht J, et al. (2013)
Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 140: 4982-4987.
8. Blitz IL, Biesinger J, Xie X, Cho KW (2013) Biallelic genome modification in F0 Xenopus tropicalis embryos using the CRISPR/Cas system. Genesis 51: 827-834.
9. Blackburn PR, Campbell JM, Clark KJ, Ekker SC (2013) The CRISPR system--keeping zebrafish gene targeting fresh. Zebrafish 10:116-118.
10. Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, et al. (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380-1389.
11. Burgess DJ (2014) Technology: Characterizing CRISPR off-target effects. Nat Rev Genet 15: 5. 12. Wu Y, Liang D, Wang Y, Bai M, Tang W, et al. (2013) Correction of a Genetic Disease in Mouse via Use of CRISPR-Cas9. Cell Stem Cell 13:
659-662.
13. Ebina H, Misawa N, Kanemura Y, Koyanagi Y (2013) Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep 3: 2510.14. Bloom K, Ely A, Mussolino C, Cathomen T, Arbuthnot P (2013) Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol Ther 21:
1889-1897.
15. Iiizumi S, Kurosawa A, So S, Ishii Y, Chikaraishi Y, et al. (2008) Impact of non-homologous end-joining deficiency on random and targeted DNA integration: implications for gene targeting. Nucleic Acids Res 36:6333-6342. 16. Zhou J, Shen B, Zhang W, Wang J, Yang J, et al. (2014) One-step generation of different immunodeficient mice with multiple gene
modifications by CRISPR/Cas9 mediated genome engineering. Int J Biochem Cell Biol 46: 49-55.
17. Cho SW, Kim S, Kim JM, Kim JS (2013) Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 230-232.
Science 315: 1709-1712.
2. Richter H, Randau L, Plagens A (2013) Exploiting CRISPR/Cas:interference mechanisms and applications. Int J Mol Sci 14: 14518-14531.
3. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, et al. (2012) Aprogrammable dual-RNA-guided DNA endonuclease in adaptive bacterial
immunity. Science 337: 816-821.
4. Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, et al. (2013)Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31: 227-229.
5. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, et al. (2013)High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31: 822-826.
6. Cradick TJ, Fine EJ, Antico CJ, Bao G (2013) CRISPR/Cas9 systemstargeting beta-globin and CCR5 genes have substantial off-target activity.Nucleic Acids Res 41: 9584-9592.
7. Hruscha A, Krawitz P, Rechenberg A, Heinrich V, Hecht J, et al. (2013)
Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 140: 4982-4987.
8. Blitz IL, Biesinger J, Xie X, Cho KW (2013) Biallelic genome modification in F0 Xenopus tropicalis embryos using the CRISPR/Cas system. Genesis 51: 827-834.
9. Blackburn PR, Campbell JM, Clark KJ, Ekker SC (2013) The CRISPR system--keeping zebrafish gene targeting fresh. Zebrafish 10:116-118.
10. Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, et al. (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380-1389.
11. Burgess DJ (2014) Technology: Characterizing CRISPR off-target effects. Nat Rev Genet 15: 5. 12. Wu Y, Liang D, Wang Y, Bai M, Tang W, et al. (2013) Correction of a Genetic Disease in Mouse via Use of CRISPR-Cas9. Cell Stem Cell 13:
659-662.
13. Ebina H, Misawa N, Kanemura Y, Koyanagi Y (2013) Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep 3: 2510.14. Bloom K, Ely A, Mussolino C, Cathomen T, Arbuthnot P (2013) Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol Ther 21:
1889-1897.
15. Iiizumi S, Kurosawa A, So S, Ishii Y, Chikaraishi Y, et al. (2008) Impact of non-homologous end-joining deficiency on random and targeted DNA integration: implications for gene targeting. Nucleic Acids Res 36:6333-6342. 16. Zhou J, Shen B, Zhang W, Wang J, Yang J, et al. (2014) One-step generation of different immunodeficient mice with multiple gene
modifications by CRISPR/Cas9 mediated genome engineering. Int J Biochem Cell Biol 46: 49-55.
17. Cho SW, Kim S, Kim JM, Kim JS (2013) Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 230-232.



