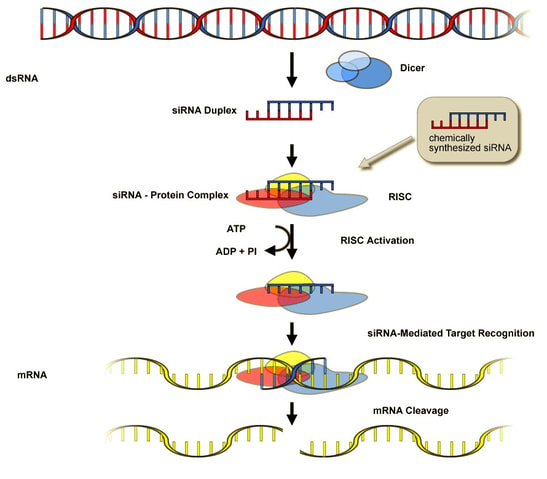
RNAi實驗原理
RNA干擾(RNA interfering,RNAi)現象是由與靶基因序列同源的雙股RNA(double-stranded RNA,dsRNA)引發的廣泛存在於生物體內的序列特異性基因轉錄後的沉默過程。細胞中的核糖核酸酶III家族成員之一的,dsRNA專一性的核酸酶Dicer將dsRNA裂解成由21-25個核苷酸組成的小干擾RNA (small interfering RNA,siRNA),隨後siRNA作為介導子引起專一性地降解相同序列的mRNA,從而阻斷相應基因表達的轉錄後基因沉默機制。
siRNA 合成服務 (合成常見問題請參考下方Q&A)
如何在細胞株中抑制基因的表達 ?
使用合成的siRNA干擾核酸物質 Common siRNA Oligo (一般情形使用, 屬於第一代 標準型siRNA)
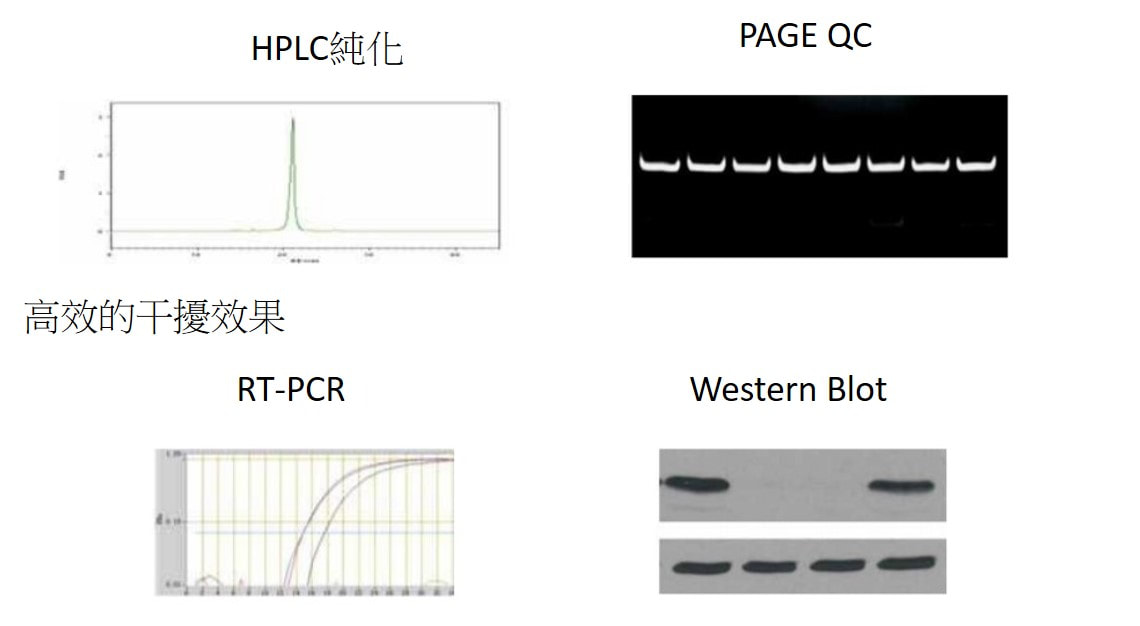
HPLC純化,純度大於97%,100%除去尚未配對的單鏈,品質優良 ISO9001,價格便宜,只需用包裝盒內的通用緩衝溶液稀釋後即可,可根據客戶提供的每條基因設計四個靶點的siRNA(費用另計 合成費用*4)。即可單獨使用又可隨意組合,以“ siRNA Pool ”形式提高抑制效率。
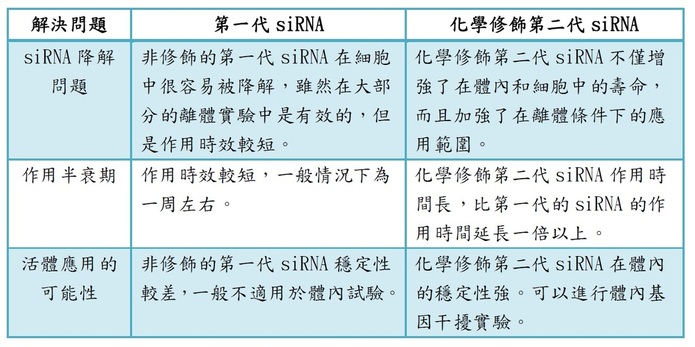
化學修飾 siRNA Oligo (可以增加 siRNA作用時間, 屬於第二代siRNA)
RNAi技術的最大難題就是化學合成siRNA穩定性問題。化學修飾siRNA最大的目標就是一次性獲得最佳的實驗資料。我們的方法就是使用RNAi轉染試劑將化學修飾siRNA導入哺乳動物細胞中,獲得如下滿意結果:1. 高效的基因沉默,2. 高穩定性—在血清和培養基中,3. 最大限度減少副作用,4. 更長的作用時間。
使用合成的siRNA干擾核酸物質 Common siRNA Oligo (一般情形使用, 屬於第一代 標準型siRNA)
HPLC純化,純度大於97%,100%除去尚未配對的單鏈,品質優良 ISO9001,價格便宜,只需用包裝盒內的通用緩衝溶液稀釋後即可,可根據客戶提供的每條基因設計四個靶點的siRNA(費用另計 合成費用*4)。即可單獨使用又可隨意組合,以“ siRNA Pool ”形式提高抑制效率。
化學修飾 siRNA Oligo (可以增加 siRNA作用時間, 屬於第二代siRNA)
RNAi技術的最大難題就是化學合成siRNA穩定性問題。化學修飾siRNA最大的目標就是一次性獲得最佳的實驗資料。我們的方法就是使用RNAi轉染試劑將化學修飾siRNA導入哺乳動物細胞中,獲得如下滿意結果:1. 高效的基因沉默,2. 高穩定性—在血清和培養基中,3. 最大限度減少副作用,4. 更長的作用時間。
siRNA 標準型合成價格表 (合成常見問題請參考下方Q&A)
編號 |
產品名稱 |
規格 |
純化方式 |
價格 |
交貨期 |
ZGGA01001 |
siRNA合成 |
2 OD |
HPLC |
5500 |
10個工作天 |
ZGGA01002 |
siRNA合成 |
3 OD |
HPLC |
6000 |
10個工作天 |
ZGGA01003 |
siRNA合成 |
4 OD |
HPLC |
6500 |
10個工作天 |
ZGGA01004 |
siRNA合成 |
5 OD |
HPLC |
7000 |
10個工作天 |
ZGGA01005 |
siRNA合成 |
6 OD |
HPLC |
8000 |
10個工作天 |
ZGGA01006 |
siRNA合成 |
7 OD |
HPLC |
9000 |
10個工作天 |
ZGGA01007 |
siRNA合成 |
8 OD |
HPLC |
10000 |
10個工作天 |
ZGGA01008 |
siRNA合成 |
10 OD |
HPLC |
12000 |
10個工作天 |
另有大量包裝,請洽詢
本公司提供的siRNA相關產品直接作用於靶基因mRNA,因此我們建議您在收到我們的相關產品後先進行實時定量PCR檢測,以確定靶基因mRNA表達水平的變化,進而直接確認我們合成或建構的siRNA產品是否有效。
備註:保證序列設計符合設計原則,保證序列合成和訂單要求一致,保證質量和純度達到共同保證要求,不保證干擾效果。非承諾質量問題,無效也需付款。
所有產品僅限用於科研實驗,僅針對細胞水平,體內效果由於影響因素較多,不做任何體內效果保證。
以上報價為一條合成的報價,如有其他需求請mail詢問。
siRNA 化學修飾價格表(報價不含siRNA合成價格)
末端修飾方式 |
規格 |
純化方式 |
價格 |
5' FAM |
2 OD |
HPLC |
3000 |
5' Biotin |
2 OD |
HPLC |
3000 |
5' Amine |
2 OD |
HPLC |
3000 |
5' Phosphate |
2 OD |
HPLC |
3000 |
5' Thiol |
2 OD |
HPLC |
3000 |
5' Cholesteroi |
2 OD |
HPLC |
3000 |
鹼基修飾方式 |
規格 |
純化方式 |
價格 |
Deoxyinosine |
2 OD |
HPLC |
1500 |
2' Fluoro dU |
2 OD |
HPLC |
1500 |
2' Fluoro dC |
2 OD |
HPLC |
1500 |
2' OMe rU |
2 OD |
HPLC |
1500 |
2' OMe rC |
2 OD |
HPLC |
1500 |
2' OMe rG |
2 OD |
HPLC |
1500 |
2' OMe rA |
2 OD |
HPLC |
1500 |
siRNA 螢光修飾價格表 Fluorescent Dye-labeled siRNA Oligo
編號 |
產品名稱 |
規格 |
純化方式 |
價格 |
交貨期 |
ZGA03004 |
Fluorescent dye labeled siRNA |
4 OD |
HPLC |
9000 |
10個工作天 |
ZGA03005 |
Fluorescent dye labeled siRNA |
5 OD |
HPLC |
10000 |
10個工作天 |
ZGA03010 |
Fluorescent dye labeled siRNA |
10 OD |
HPLC |
18000 |
10個工作天 |
ZGA03025 |
Fluorescent dye labeled siRNA |
25 OD |
HPLC |
35000 |
10個工作天 |
siRNA 合成服務 Q & A
1. siRNA的相關的一些技術數據及注意事項
(1)的siRNA的平均分子量13300。
(2)siRNA oligo的OD,nmol和質量間有相應的公式可以計算;一般情況下,對於一個21 bp的siRNA oligo,有如下簡單關係:1 OD duplex = 3.0 nmols = 40 ug
(3)1 OD's siRNA欲溶解為20 uM的樣品,可用150 uL DEPC H2O去重懸1 OD's siRNA,溶解後為20 uM的樣品。
(4)由於Oligo RNA呈很輕的乾粉狀附在管壁上,打開時極易散失,所以打開管子前先離心,然後再慢慢打開管蓋,溶解時請加足量DEPC水後蓋上管蓋,振盪溶解。
(5)螢光標記的RNA,如FAM,HEX,TAMRA等標記,因為對光敏感,必須避光保存。
2.哺乳動物siRNA設計需要注意的幾個方面
(1)從轉錄本(mRNA)的AUG起始密碼開始,尋找“AA”二連序列,並記下其3'端的19個鹼基序列,作為潛在的siRNA靶位點。正向鏈和反向鏈都採用這19個鹼基(不包括AA重複)來設計。
(2)避免在起始密碼子或無義區域附近選擇目的序列。
(3)siRNA序列的GC含量應為30%-60%左右。
(4)在設計siRNA時不要針對5'和3'端的非編碼區(untranslated regions,TRs),原因是這些地方有豐富的調控蛋白結合區域,而這些UTR結合蛋白或者翻譯起始複合物可能會影響siRNP核酸內切酶複合物結合mRNA從而影響siRNA的效果。
(5)將挑選的序列在公共資料庫中進行比較以確保目的序列與其它基因沒有同源性。
(6)將潛在的序列和相應的基因組資料庫(人,或者小鼠,大鼠等等)進行比較,排除那些和其他編碼序列/EST同源的序列。例如使用BLAST(www.ncbi.nlm.nih.gov/BLAST/)。
(7)選出合適的目標序列進行合成。通常一個基因需要設計多個靶序列的siRNA,以找到最有效的siRNA序列。
(8)陰性對照:一個完整的siRNA實驗應該有陰性對照,作為陰性對照的siRNA應該和選中的siRNA序列有相同的組成,但是和mRNA沒有明顯的同源性。通常的做法是將選中的siRNA序列打亂,同樣需要檢查它和其他基因是否具有同源性。
(9)有結果顯示,UU結尾和dTdT結尾的siRNA在效果上沒有區別,因為這個突出端無需和靶序列互補。合成siRNA時可直接提供以AA打頭的21個鹼基序列。
Q: human u6 promoter 表達框,是如何設計的?
human U6 promoter表達框包括human U6 promoter、終止碼TTTTT和中間自行設計的抑制序列或抑制序列的髮卡結構。從人基因組中可以擴增出U6 promoter,後面的序列全部自己設計。
Q: 一般設計多少bp的siRNA,以及轉錄的終止子?
因為人工合成siRNA價格太高,現在經常採用RNAi expression vector,抑制序列一般在19-22bp。使用Pol III啟動子時轉錄終止碼為5'-TTTTT-3',而使用Pol II啟動子則為polyA。其實這兩種終止碼都不能完全避免通讀(readthrough)現象的發生。現在RNAi多用前者,是因為其較短,不形成複雜的空間結構;後者因為較長,形成的空間結構可能會對抑制序列髮卡結構的形成構成空間阻礙或產生遮擋效應。
siRNA 對照
A.普通陰性對照
可提供與目的基因序列無同源性的通用陰性對照和將選中的siRNA序列打亂(scrambled)的普通陰性對照。
(1)siRNA實驗應該有陰性對照;
(2)通用陰性對照為與目的基因的序列無同源性的普通陰性對照;
(3)Scrambled陰性對照和選中的siRNA序列有相同的組成,但是和mRNA沒有明顯的同源性;
(4)陰性對照需要確定和目的靶細胞中其它基因沒有同源性。
B.螢光標記陰性對照
可提供與目的基因序列無同源性的螢光標記(6-FAM)通用陰性對照。
(1)RNAi negative control與哺乳動物基因無同源性;
(2)通過標記螢光,可以方便地在螢光顯微鏡下觀察轉染情況
(3)可用於優化轉染條件和評價轉染效率;
(4)具備很好的pH耐受性,在活細胞中更穩定
C.siRNA陽性對照
陽性對照作為一個實驗系統檢查是很重要的。您可以利用陽性對照來確認RNAi實驗中轉染、RNA提取和基因表達檢測方法是可靠的。
(1) LaminA/C
(2) GFP22
(3) Luciferase GL2
(4) MAPK1
(5) Beta-Actin
(6) Vimentin
(7) P53
(8) GAPDH
(9) Cyclophilin B
siRNA轉染
A.siRNA轉染的方法
哺乳動物轉染的常見方法有:磷酸鈣共沉澱、電穿孔法、DEAE-葡聚糖和polybrene、機械法(例如,顯微注射和基因槍)、陽離子脂質體試劑,其中陽離子脂質體試劑轉染法是目前最常用的轉染方法。
應用脂質體型轉染試劑進行轉染需要注重的幾個方面:
1.轉染試劑的用量
2.siRNA的用量
3.轉染時的細胞密度
4.轉染時的操作順序
5.細胞與轉染試劑/siRNA複合物的溫浴的時間
B . Lipofectamin2000 轉染試劑
適合的siRNA(DNA):lipofetamin2000比例對核酸的高效轉染有重要影響;我們推薦的DNA:lipofetamin2000為1:0.5—1:5(ug:ul),siRNA:lipofectamin2000為1:0.01-1:0.1(pmol:ul)一般情況下,此範圍內都可獲得高的轉染效率。
選擇最適合的轉染試劑和轉染條件,往往取決於不同的哺乳動物細胞類型和不同的核酸分子。
Lipofectamin2000適用於核酸的體內和體外操作,可應用於DNA、RNA、反向寡核苷酸、siRNA的轉染,也可應用於DNA/siRNA的共轉染操作;是一種新型的高效siRNA轉染試劑。
Lipofectamin2000的應用領域:
□ 原代培養細胞和轉化細胞株的基因轉染
□ siRNA高通量轉染試驗
□ DNA轉染;DNA和siRNA的共轉染
□ 核酸(siRNA、DNA、RNA)的體內導入試驗
□ 貼壁細胞和懸浮細胞轉染
細胞培養用品 ‖ siRNA/DNA ‖ 培養基最終體積 ‖ Lipofectamin2000(siRNA/DNA)
96孔板 ‖ 5pmol/0.2μg ‖ 100Μl ‖ 0.25μl/0.5μl
24孔板 ‖ 20pmol/0.8μg ‖ 500μL ‖ 1μl /2μl
12孔板 ‖ 40 pmol /1.6μg ‖ 1 mL ‖ 2μl /4μl
6孔板 ‖ 100 pmol /4.0μg ‖ 2 mL ‖ 5μl /10μl
35 mm ‖ 100 pmol /4.0μg ‖ 2 mL ‖ 5μl /10μl
60 mm ‖ 600 pmol /8.0μg ‖ 5 mL ‖ 10μl /20μl
C . Lipofectamin2000適用的細胞類型
Lipofectamin2000轉染試劑可廣泛應用於多種細胞系的DNA和siRNA轉染如:HeLa(人頸部癌細胞)、MCF-7(人乳房癌細胞)、Hep3B(人肝細胞癌細胞)、COS-7(猴腎細胞)、Neuro-2a(鼠神經母細胞瘤細胞)、NIKS(人角質化細胞)、B16(鼠黑素瘤細胞)、DLD-1(人結腸癌細胞)、NIH/3T3(鼠胚胎成纖維細胞)、HT-29(人結腸腺癌細胞)、A549(人肺癌細胞)、CHO-k1(倉鼠卵巢細胞)和293(腺病毒5 DNA轉化的人胚胎腎細胞),SVRbag4細胞等。
D.轉染前細胞培養
細胞培養用品/表面積(mm2/孔)/細胞密度/培養基(μL /孔)
96孔板 / 50 / 1.5×104-5.0×104 / 100μL
48孔板 / 100 / 3.0×104-1.0×105 / 200μL
24孔板 / 200 / 8.0×104-2.0×105 / 500μL
12孔板 / 401 / 1.6×105-4.0×105 / 1.0 mL
6孔板 / 962 / 3.0×105-8.0×105 / 2.0 mL
35 mm / 962 / 3.0×105-8.0×105 / 2.0 mL
60 mm / 2827 / 1.0×106-2.5×106 / 6.0 mL
F.貼壁細胞轉染程式
選用生理狀態良好的細胞對提高轉染效率很
重要。siRNA(DNA)和lipofectamin的用量和兩者的比例可在推薦範圍內適當調整。
1.轉染前一天,4-5×104細胞接種在24孔板上, 0.5mL含FBS和抗生素的DMEM(或Opti-MEM,其他培養基)細胞培養基。
2. 選擇用於初期接種的細胞數量,應能在24小時內使細胞匯合達到70-90%。
3.在50μl的DMEM(或Opti-MEM,或其他無血清培養基)無血清培養基加入20pmol siRNA(或0.8μg DNA),柔和混勻
4.混勻lipofectamin試劑,用50μl無血清的DMEM或Opti-MEM,或其他無血清培養基)稀釋1μl lipofectamin試劑(DNA轉染時,則加入2μllipofectamin試劑),輕輕混勻,室溫放置5分鐘
5.將稀釋好的siRNA和RNAi-Mate試劑混合;輕柔混勻,室溫放置20分鐘,以便形成siRNA/lipofectamin(或DNA/lipofectamin)複合物
6. 將100μl siRNA/lipofectamin(或DNA/lipofectamin)複合物加到含有細胞和培養基的培養板的孔中,來回輕柔搖晃細胞培養板板
7. 細胞在CO2培養箱中37℃溫育24h-48h後,進行轉染後的其它檢測步驟。如果細胞株比較敏感,孵育4-6小時後,除去複合物,更換培養基。
G.siRNA體內導入方法
1. 適量的siRNA或DNA溶於不含RNA酶的無菌水中,輕輕混勻,因為注射液體積有限,建議採用高濃度的siRNA或DNA,一般DNA為2μg /μL、siRNA為10μg /μL。
2. 取適量的DNA、siRNA或siRNA\DNA複合物與lipofectamin混合。例如,在1#管中加入0.5μL的DNA(1μg)和0.5μL的siRNA(5μg),在2#管中加入0.55μL的lipofectamin(24μg)和0.45μL不含RNA酶的無菌水中,將1#管中的溶液加入2#管中,在室溫下溫育30分鐘,以形成siRNA/DNA-lipofectamin複合物。
3.製備的siRNA/DNA-lipofectamine複合物可用於體內導入siRNA、DNA或siRNA\DNA
siRNA轉染常見問題與建議
A . 轉染效率低
Q: RNAi-Mate:siRNA(DNA)未優化
建議:我們推薦的DNA:lipofetamin2000為1:0.5—1:5(ug:ul),siRNA:lipofectamin2000為1:0.01-1:0.1(pmol:ul)
Q: siRNA/lipofectamin(或DNA/lipofectamin)複合物濃度低
可適當提高siRNA/lipofectamin(或DNA/lipofectamin)複合物的濃度
Q:細胞生長狀態不好
建議:非最佳狀態的細胞會降低轉染效率,建議細胞接種後在24小時內達到70-90%,並在24小時內完成轉染
Q:DNA或siRNA的純度太低
建議:使用高品質的DNA或siRNA,最好使用柱純化的DNA和HPP級的siRNA
Q:稀釋DNA或siRNA的培養基中含有血清
建議:一般情況下,血清不會明顯降低siRNA/lipofectamin(或DNA/lipofectamin)複合物的形成,建議使用不含血清的培養基稀釋DNA或siRNA
B . 實驗的重複性不好
Q: 細胞匯合情況不一致
建議:使用相同數量的種細胞,接種後的培養時間、培養條件一致
Q: 細胞傳代次數太多
建議: 使用傳代次數較低的細胞
Q: 細胞出現明顯死亡
建議:與細胞生存相關的關鍵基因被關閉重新設計實驗
Q:細胞狀態欠佳
建議:使用傳代次數較低的細胞;細胞接種後在24小時內達到70-90%,並在24小時內完成轉染。
Q: siRNA/RNAi-Mate(或DNA/lipofectamin)複合物濃度過高
建議: siRNA/lipofectamin(或DNA/lipofectamin)複合物一般不會影響細胞生長,當濃度過高時,有時也會產生毒性。
C . 常見問題:
Q:力鈞提供的siRNA是雙鏈的還是單鏈的?
力鈞的siRNA是雙鏈的,而且也是按照雙鏈來定價的。
Q:我們需要提供什麼資訊用於siRNA的合成?
您需要準確地提供siRNA的19個核苷酸的靶序列和懸垂的合成物,或者您也可以提供相應地基因的GeneID或者Accession Number,
Q:如何選擇siRNA的懸垂組成?
懸垂的序列組成是由顧客自行選擇的。最近研究表明懸垂的組成在mRNA靶識別和酶解中不是很重要。懸垂可能在形成RISC的過程中起結構的作用。很多研究人員選擇dTdT是因為研究表明去氧核糖核苷能保護siRNA免受酶解。合成序列最重要的是確定靶mRNA的19個鹼基的核心結構。
Q:針對人體基因設計的siRNA對其他物種是否也有效?
一般siRNA都具有物種專一性,很少與其他物種有相同的靶位點,所以針對人體基因設計的siRNA不會沉默其他物種的同源序列。然而,也有研究表明siRNA經過專一性設計後能對兩個或兩個以上的物種有效,這需要仔細進行siRNA設計和生物資訊學分析。
Q:你們提供的siRNA是怎樣裝運的?如果在常溫下放置了一個星期還有效嗎?
力鈞為您提供的siRNA是凍乾粉包裝的,在常溫下運輸。這些凍乾的樣品在室溫下能穩定保存2-4個星期,所以放置一個星期不會影響其沉默效果。但我們建議您收到樣品後最好保存於-20℃或-70℃冰箱中。
Q:在體外實驗中,需要多少量的siRNA?
我們建議您用於實驗的siRNA的濃度為100nM。1nmol siRNA的量對於一個24孔板或是96孔板的實驗已經足夠了。用100nM 的siRNA轉染時只得到50%沉默效率,我可以把siRNA的濃度增加到200nM甚至400nM嗎?增加siRNA的濃度一般不能改進沉默效率。高濃度的siRNA將可能導致去靶作用和對細胞的毒性。siRNA的高基因沉默效率來自於合理的設計,在100nM 甚至更低的濃度都可能有75%的沉默效率。另外,低的轉染效率會導致低的沉默效率,建議您更進一步優化siRNA的導入條件。
Q: cDNA產量的很低
可能的原因:
1. RNA範本品質低
2. 對mRNA濃度估計過高
3. 反應體系中存在反轉錄酶抑制劑或反轉錄酶量不足
4. 同位素磷32過期
5. 反應體積過大,不應超過50μl
Q: 定量RNA的公式是什麼?
研究者可以用Beer法則定量RNA:吸光度(260nm)=(摩爾消光係數)*(濃度)*(路徑長度,cm)。為了便於理解,等式變為:濃度=(吸光度,260nm)/[(摩爾消光係數)*(路徑長度,cm)]。當使用一個標準的10mm比色皿時,在公式中路徑長度這個變數等於1。
Q: 6uL濃度為10uM的siRNA溶解液中含有多少ug的siRNA?
首先計算含有多少nmol的siRNA:
a. 等式: ? nmol = (6 uL)(10 umol/L);b. 單位換算: ? nmol = (6 uL)(10 umol/L)(1 L/1,000,000 uL)(1,000 nmol/umol);c. 答案: ? nmol = 0.06 nmol。
然後利用siRNA的平均分子量(13,300 g/mol)把nmol變為ug:
a. 等式: ? ug = (0.06 nmol)(13,300 g/mol);b. 單位換算: ? ug = (0.06nmol)(13,300 g/mol)(1mol/1,000,000,000 nmol)(1,000,000ug/g);c. 答案: ? ug = 0.798≈0.8 ug。所以6uL濃度為10uM的siRNA溶解液中含有0.8ug的siRNA。
Q: 我需要濃度為20uM的樣品,如何計算重懸siRNA緩衝液的量?
樣品濃度的計算如下:(siRNA的量,nmol)/(重懸體積,uL)=樣品濃度,umol/L。在解答前應先統一單位,確保單位可以抵消。例如:您購買了20nmol的siRNA,想溶解為50uM的樣品。可按以下方法計算重懸緩衝液體積:a. 等式: (20 nmol)/ ? uL = 50 umol/L;b. 解答未知量: ? uL = (20 nmol)(1 L/50 umol);c. 單位換算: ? uL = (20 nmol)(1 L/50 umol)(1 umol/1,000 nmol)(1,000,000 uL/1 L);d.答案: ? uL = 400 uL。因此,應該使用400uL緩衝液去重懸20nmol的siRNA,溶解後為50uM的樣品。
Q: 一定需要陰性對照嗎?
是,陰性對照是RNA干擾實驗中不可缺少的。由於在超過200 nM的濃度下,siRNA有可能會導致非專一性的壓力反應,在實驗體系中必需設置陰性對照。它能夠幫助我們確認基因表達水準的降低是否是序列專一性的RNAi的結果。由於siRNA的合成方法和工藝以及轉染試劑等因素可能導致廣泛的基因沉默現象。如果沒有陰性對照,研究人員很可能錯誤地將廣泛的、非專一性基因沉默當作由RNAi引起的基因專一性沉默。
Q: 常用的陰性對照有哪些類型?
常用的陰性對照大體分為兩種,一種是使用通用陰性對照序列,該序列已經被上千篇文章使用;另一類是使用和靶基因siRNA打亂序列的siRNA作為陰性對照,這樣的陰性對照和通用序列相比較,一方面合是按照定制產品價格合成的,另一方面,由於打亂序列是沒有經過驗證的序列,有可能會產生off-target現象。因此,除非有非常特殊的要求,最好使用通用序列作為陰性對照。
Q: 在陰性對照體系中和實驗體系中觀察到同樣的實驗結果,這是什麼原因?
這個結果充分說明了設置陰性對照的必要性。該結果表明您所觀察到的表型不是序列專一性knockdown產生的表型,您需要降低siRNA的工作濃度。
Q: 為什麼說陽性對照在RNA干擾實驗中很重要?
陽性對照作為一個實驗系統檢查是很重要的。也就是說,當您看到siRNA陽性對照的預期實驗結果時,您能確保在您的實驗方法中您的轉染、RNA提取物和檢測方法是可靠的。通常最好的陽性對照是內在的對照。
Q: 你們能提供預先合成的siRNA對照麼?
可以。力鈞能提供許多預先合成的用於RNA干擾實驗的對照雙鏈。
Q: 用於對照的siRNA的最佳濃度是多少?
陽性對照和陰性對照的siRNA的濃度都應該與基因專一性的siRNA的濃度相同。
Q: 在siRNA 上進行標記的最佳位元點在哪裡?
反向鏈的5'端標記會影響siRNA的沉默活性,所以不推薦標記這個位點。在其他三個末端進行標記對沉默活性幾乎沒有影響。有研究表明正向鏈的5'端標記是最有效的化學合成位元點。
Q: 螢光標記的siRNA是不是可以作為良好的對照?如何檢測螢光標記的siRNA?
已經有為數不少的實驗室研究過螢光標記的siRNA的螢光檢測結果和knockdown效率之間的正相關關係。螢光標記雙鏈是最常用的優化轉染條件的方法。可以用流式細胞儀或者螢光共聚焦顯微鏡檢測標記的siRNA。
Q: 螢光素的最大吸收率和發射率各在哪個波段?
螢光素在495nm處有最大吸收率,在520nm處有最大發射率
Q: 如何篩選轉染試劑?
好的轉染試劑有以下特點:1、對siRNA有較高的轉染率;2、對細胞的毒性小。在mRNA水準檢測siRNA導入的效果比用看家基因的siRNA陽性對照檢測更為準確。
Q: 轉染過程中發現大量細胞死亡,我應該如何處理?
如果出現細胞大量死亡,意味著您的轉染條件仍然需要優化:
轉染條件的優化一般包括以下幾個方面:
1. 調整轉染試劑的濃度;
2. 在轉染後適當的時間內更換無轉染試劑的培養液,
3.調整細胞的生長狀態;一般處於良好生長狀態的細胞對轉染試劑就有更好的耐受性;
4.調整轉染試劑和siRNA的比例;
如果同一管轉染試劑在不同實驗中對細胞的毒性有差異,一般說來應該是實驗過程本身帶來的差異;
如果已經做了以上幾項工作,轉染效率仍然得不到提高,建議您更換一種轉染試劑。
Q: 如何將siRNA導入到細胞內?
使用什麼樣的轉染方法,很大程度上取決於您使用的細胞系:
1.貼壁的、易轉染的細胞,我們推薦使用RNAi-mate;
2.懸浮的或原代的細胞,我們推薦使用電擊轉化方法;
3.電擊轉化效率仍然很低的細胞,需要選擇載體系統。
Q: 我的課題主要是研究細胞凋亡相關基因,我手上有一些新基因需要使用RNA干擾進行深入研究,我該如何選擇對照系統?
用RNAi進行pathway研究,是RNAi主要應用之一。在這樣的課題中,對照系統的選擇尤其重要。已經發表的和已經驗證的siRNA為RNA干擾研究提供了系統性對照。用RNAi進行細胞凋亡相關基因的研究。
Q: 我剛剛開始接觸RNA干擾技術,從文獻上看,不同細胞應該選用不同的轉染試劑,不同的研究目的,應該使用不同的siRNA,我的經費有限,如何有效確定我的研究體系?
在開始RNA干擾研究之初,需要確定以下幾個方面:
使用化學合成的siRNA還是使用載體構建shRNA?我們推薦您使用化學合成的方法來篩選有效的目的片段,然後把您篩選出來的目的片段插入到載體,進行抗性篩選,得到穩定表達的細胞株,再做長期研究。
Q: 反向核酸和RNAi有何差異?
反向核酸和RNAi從作用原理到使用的範圍都有很大差異。這裡只能做一個簡單的介紹:從原理上說,反向核酸是一段與靶基因配對的單鏈DNA或類似DNA的片段與靶基因結合,結果阻止靶基因的轉錄或是翻譯,以往的研究表明在反向核酸的研究中,序列的有效性和有效序列的非專一性往往有比較高的正相關性,這就在一定程度上限制了反向核酸的應用。
RNAi的作用機理是雙鏈的RNA進入細胞內,導致靶基因的切割和降解。siRNA的序列可以選擇在高度特異針對靶基因的位置,也就為RNAi作為藥物研究提供了高效、低副作用的空間。自從RNAi技術問世以來,國外很多專業從事反向核酸研究的紛紛轉向RNAi研究, ISIS是一個非常就有代表性的例子。他們認為,如果使用反向核酸可以進行的研究,以及反向核酸可以涉及的研究領域,RNAi均可以毫不示弱的開展,並且應該會得到更加令人滿意的結果。
Q: 我使用合成的雙鏈DNA克隆載體,但是定序結果不正確,是DNA合成的問題,還是其他問題?
首先,品質良好的、並且是退火狀態良好的dsDNA是克隆成功的關鍵因素。一般情況下,需要挑不止一個克隆定序(通常是2個以上),如果兩個克隆的序列都是錯誤的,並且錯誤的鹼基是相同的,那麼基本可以確定是DNA oligo合成的問題. 如果兩個克隆的不同鹼基發生錯誤,有可能是合成片段本身的問題,也有可能是克隆過程造成的問題,可以再多挑1~2個克隆定序確證。大多數情況下,兩個陽性克隆中,一般至少會有一個克隆是正確的,這種個別鹼基在個別分子中的錯誤是合成和克隆過程共同造成的,但一般發生的幾率較低。
Q: 我計畫使用RT-PCR檢測基因knockdown結果,之前應該注意哪些問題?
我們推薦RT-PCR的引子應該設計在target位置的兩側,而不是同側。目前已經知道的siRNA介導的基因knockdown機制是RSIC先介導靶mRNA的切割,切割導致靶mRNA的降解,由於切割和降解可能具有不同的時間點,因此,設計於同側的PCR引子可能導致假陰性結果或knockdown效率的低估。
另外,RT-PCR結果會因為靶基因mRNA降解時間不同、細胞內靶基因mRNA豐度不同而導致假陰性結果,特別對於豐度較高的基因,推薦使用的檢測方式包括定量PCR、Northern檢測、western檢測等。這些方法會更加真實的反映RNAi的結果。
Q: 開始體內實驗前需要注意什麼問題?
體內實驗設計包含動物模型的選擇、給藥途徑、劑量和給藥次數等等。siRNA使用的數量及濃度主要取決於給藥靶點的性質,諸如腫瘤的類型,組織的類型,靶基因表達水準,動物模型個體大小等等,針對不同的研究模型,最好先查閱相關資料。
Q: 在體內研究中,最好的給藥途徑是什麼?給藥頻率是多少?
寡核苷酸可以通過大劑量給藥或使用ALZET微小泵持續給藥。大劑量給藥時要慎重,因為已有研究表明:一些毒性與寡核苷酸反向鏈的濃度有關,快速給藥時可能會導致動物下肢癱瘓或致死,所以給藥時要注意觀察。很多已發表的論文實驗是通過尾靜脈給藥的。任何給藥方式都需要優化,以確保最佳的導入方式和動物的健康。一般拿靜脈注射來說,每天注射一到兩次,連續注射一到兩周。
Q: 力鈞合成的siRNA是否可以用於動物水準的實驗?
力鈞的siRNA經過嚴格HPLC純化,已經被使用者通過局部注射用於小鼠的體內實驗,並且得到滿意的實驗結果。
Q: 小鼠體內實驗需要多少siRNA?
迄今為止還沒有明確的siRNA體內實驗使用量的計算方式,在實驗前最好先從文獻中查詢是否已經有相關的文章發表。對於常規實驗,一般使用100 μL的注射體積,濃度為10 to 500 μM。原先用antisense的研究表明,當劑量大於20 mg/kg/day (416 mM)時,會觀察到明顯的毒性。最好針對您的實驗繪製劑量反應曲線。 常規情況下,給藥劑量可以以 ~5mg/kg/day [~7.7 nmol/day or 100 mM per day]作為優化起點。需要注意的是,這個劑量只是一個預實驗的起點,最後的給藥劑量取決於動物模型、靶基因、靶組織和給藥方式等因素。
Q: 沉默效果不理想,應該如何處理?
最常見的影響沉默效果的兩個原因是:轉染效率低和siRNA序列設計的效果不理想。如果您初次使用siRNA或採用了新的細胞系,並發現沉默效果不佳,我們建議您對轉染效率進行檢測,並選擇優化轉染條件。如果您已經對實驗轉染條件進行優化但是問題依然存在,我們建議您換用另一種轉染試劑或是採用其他技術,這也許能提高轉染效率。如果已經提高了轉染效率但是沉默效果仍然未達到要求,可能是因為siRNA序列設計的效果不理想。
Q: 如何使用Pol III啟動子製備RNAi表達載體?
目前發表的RNAi表達載體大多採用3型Pol III啟動子,如human/mouse U6啟動子等。其中人U6啟動子有幾個明顯的特點:1.具有TATA box,位於-30~-25bp,被Pol III轉錄;2.在轉錄起點上游-66~47bp處存在PSE(proximal sequence element),該元件是snRNA啟動因數蛋白複合物的結合位點;3.在-244~-214存在DSE(distal sequence element);4.啟動子下游存在5'-TTTT-3'序列為Pol III提供轉錄終止信號;5.PSE和DSE之間的距離變化可顯著影響轉錄效率;6.+1位的G對轉錄效率影響較大。根據這些特點,在使用U6啟動子構建RNAi表達載體時,U6啟動子的長度最好在300bp左右,儘量不改變PSE和DSE的間距,同時保留TATA box和+1位的G,在下游應有5'-TTTTT-3'序列作為終止信號。另外可以根據需要在啟動子上游或終止信號下游設計測序引物序列、酶切位點等等。
如有興趣可以利用同尾酶設計poly-RNAi表達載體,從而可以觀察同時knockdown兩個或多個基因表達後產生的效應。若是條件允許,可以對U6啟動子進行shuffling,篩選具有特殊用途的突變的Pol III啟動子。
Q: H1啟動子和U6啟動子有什麼差別?
shRNA 載體分別使用Pol III依賴H1或者U6的啟動子。儘管H1和U6是polⅢ 型啟動子,然而可能根據使用細胞系的不同,效率有些小的差別。
Q: 是否可以插入到真核表達質體載體中?
可以,這些載體都是真核表達載體,根據國外的經驗,質體載體的knockdown作用一般僅持續一周左右,一周後被down的RNA水準即恢復原有水準,其機制尚未闡明。推薦使用逆轉錄病毒載體達到穩定轉染的效果。
Q: 為什麼使用載體導入shRNA?
在哺乳動物細胞中,RNAi可以通過直接導入特異阻斷所選擇基因表達的分子而誘導,導致產生基因功能缺失表型。這些分子可以通過化學合成、體外方法製備或者細胞內DNA範本產生。前兩種方法只能用於暫態阻斷,可能很難導入難以轉染的、不分裂的或者原代細胞類型。通過載體導入pol III 啟動子表達的短的髮夾RNA(shRNA)擴展了RNAi實驗的選擇,包括穩定和可誘導表達以及病毒導入。
如何在活體動物中抑制基因的表達 ? 使用合成的 vivo siRNA干擾核酸物質
RNAi活體研究(vivo RNAi)
雖然RNAi干擾已經在體外(in vitro)研究中成為一個強大的研究方法,但是如何在活體動物中去抑制基因表達是一個難題。為此,研究人員現在應用RNAi活體研究(vivo RNAi)的方法來進行研究。雖然此項研究還處於早期,但是活體RNAi用已經越來越廣使用並取得了不少進展
在活體動物中,可以利用局部注射或者全身性給藥的方式,利用化學修飾過的 vivo siRNA去抑制基因表達。我們的單體的修飾方式有2’-F, 2’-Ome,磷酸化修飾,膽固醇修飾,巰基修飾等。我們提供的 vivo siRNA經過離子交換式的HPLC純化,純度>95%以上。
RNAi活體研究(vivo RNAi)
雖然RNAi干擾已經在體外(in vitro)研究中成為一個強大的研究方法,但是如何在活體動物中去抑制基因表達是一個難題。為此,研究人員現在應用RNAi活體研究(vivo RNAi)的方法來進行研究。雖然此項研究還處於早期,但是活體RNAi用已經越來越廣使用並取得了不少進展
在活體動物中,可以利用局部注射或者全身性給藥的方式,利用化學修飾過的 vivo siRNA去抑制基因表達。我們的單體的修飾方式有2’-F, 2’-Ome,磷酸化修飾,膽固醇修飾,巰基修飾等。我們提供的 vivo siRNA經過離子交換式的HPLC純化,純度>95%以上。



